Publié le 25 nov 2023Lecture 8 min
Maladie de Kimura ou lymphogranulome éosinophile
Frédéric TRUFFINETa , Marie JACHIETb , Pierre ISNARDc , Thierry JO MOLINAc , Benjamin TERRIERa,d*, Paris
La maladie de Kimura est une maladie inflammatoire, historiquement décrite chez de jeunes hommes asiatiques(1). Les premières descriptions, par Kim et Szeto, remontent à 1937 dans la littérature chinoise. En 1948, le japonais Kimura donnera son nom à la maladie en venant compléter ces descriptions avec l’aspect histologique. La maladie est alors décrite par un tableau clinico-biologique associant des indurations sous-cutanées, des adénopathies de la sphère cervicale et/ou une hypertrophie de glandes salivaires, et une hyperéosinophilie.
La maladie de Kimura, plus fréquente chez les hommes avec un sex-ratio homme/ femme de 7/3, touche principalement les individus jeunes entre 25 et 40 ans(1,2). Son incidence est très rare en Occident alors qu’elle est plus fréquente en Asie, notamment en Chine et au Japon. L’étiologie est inconnue, mais des hypothèses sont émises, principalement celle d’une réaction inflammatoire chronique suite à une stimulation antigénique, notamment à Candida albicans(3).
UN DIAGNOSTIC ORL OU DERMATOLOGIQUE
Les spécialités le plus souvent confrontées à cette pathologie bénigne sont les ORL pour l’exploration d’une masse cervicale ou rétro-auriculaire ou d’une hypertrophie glandulaire, les dermatologues pour l’exploration d’une masse ou d’une induration sous-cutanée ou de lésions prurigineuses, parfois les hématologues et les internistes pour le bilan d’adénopathies, et bien sûr les médecins généralistes pour l’ensemble de ces manifestations.
La présentation clinique initiale peut varier, mais le point commun de toutes ces situations est l’absence d’altération de l’état général, contrastant avec des répercussions parfois importantes sur le plan esthétique.
La présentation clinique classique repose sur l’association de :
– nodules sous-cutanés ;
– adénopathies satellites locorégionales ;
– hypertrophie des glandes salivaires.
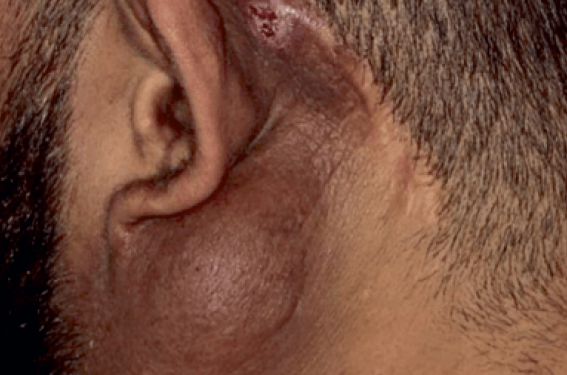
• Nodules sous-cutanés. Les nodules sont bien limités, classiquement indolores et non prurigineux. Ils siègent fréquemment dans la région cervico-céphalique, ORL, rétro-auriculaire (figure 1A), sur le cuir chevelu (figure 1B), et plus rarement sur les avant-bras (figure 1D), les cuisses, les tibias et les fesses. La progression est lente, parfois sur plusieurs années. Les lésions peuvent mesurer plusieurs centimètres, classiquement entre 3 et 10 cm, et sont multiples dans 40 % des cas. Une coloration cutanée jaune/orangée en regard est possible(4).
Des lésions cutanées non spécifiques sont rapportées dans plus de la moitié des cas. Ces lésions prurigineuses peuvent mimer une dermatite atopique, un prurigo nodulaire ou un lichen plan(4).
Figure 1. Lésions cutanées typiques au cours de la maladie de Kimura.
A. Nodule sous-cutané de localisation typique rétro-auriculaire de taille > 5 cm ; B. Nodules multiples de localisation périauriculaire et cuir chevelu ; C. Papules et nodules sur la joue ; D. papulo-nodules excoriés multiples disséminés sur le bras.
• Adénopathies satellites locorégionales. Des adénopathies loco-régionales sont quasi systématiques, classiquement dans les régions cervicales et péri-auriculaires, mais parfois sous-mammaires, axillaires ou épitrochléennes. Ces adénopathies peuvent être la seule manifestation clinique décelable de la maladie.
• Hypertrophie des glandes salivaires. Une hypertrophie des glandes salivaires, indolore, est fréquemment observée, entre 22 et 43 % selon les cohortes(1,2). Une atteinte orbitaire à l’origine d’une exophtalmie voire d’un véritable syndrome de Mikulicz(5,6) est possible dans de rares cas.
• Atteinte rénale. La complication systémique rare à connaître est le syndrome néphrotique, associé dans environ 10 % des cas. Une protéinurie est observée chez 10 à 20 % des patients correspondant dans 70 % des cas à un syndrome néphrotique(8). Ce dernier peut être présent dès le début de la maladie ou apparaître lors de son suivi. Il est donc indispensable de rechercher une protéinurie lors du diagnostic et durant le suivi tous les 6 à 12 mois. La glomérulo néphrite extramembraneuse et la glomérulonéphrite membrano-proliférative sont les atteintes les plus fréquentes(9,10), les autres lésions étant des atteintes de type lésions glomérulaires minimes, hyalinose segmentaire et focale, néphropathie à IgA et néphropathie tubulo-interstielle aiguë avec infiltration d’éosinophiles(11).
Devant une atteinte rénale, la biopsie rénale sera à discuter. Le diagnostic d’atteinte rénale spécifique de la maladie de Kimura sera retenu en fonction du contexte, en particulier la preuve de la maladie de Kimura sur le plan extrarénal et l’élimination des diagnostics différentiels. Devant une atteinte glomérulaire extramembraneuse, la recherche d’anti corps ou d’antigènes spécifiques tels que les PLA2R ou les exostosines pourra être réalisée.
• Manifestations biologiques. Une éosinophilie sanguine supérieure à 500/mm3 est fréquente, avec des valeurs variables dans la littérature (entre 80 et 100 % selon les séries)(2,4,7), associée à une augmentation quasi constante des IgE totales.
• L’absence d’altération de l’état général est un élément majeur du diagnostic, dont la présence devra faire rechercher des diagnostics différentiels.
• Imagerie. Une exploration par scanner ou TEP-scanner est souvent réalisée, notamment devant dans le bilan d’adénopathies, afin d’éliminer les diagnostics différentiels. On peut alors retrouver de multiples adénopathies locorégionales et/ou médiastinales. Ces adénopathies sont hypermétaboliques et sont isolées, sans autre lésion hypermétabolique visible au TEP-Scanner.
UN DIAGNOSTIC HISTOLOGIQUE
L’examen anatomopathologiue, à la suite d’une biopsie et/ou de l’exérèse d’une lésion (figure 2), montre un ganglion d’architecture conservée avec une hyperplasie lymphoïde folliculaire floride, une expansion des territoires inter-folliculaires avec hyperplasie des veinules postcapillaires, et une éosinophilie modérée à massive, intra, péri ou interfolliculaire formant parfois des « abcés » à éosinophiles. Une composante fibreuse est parfois présente avec une topographie capsulaire et intraganglionnaire de distribution souvent périvasculaire(12).
Figure 2. Maladie de Kimura : aspect histologique de la lymphadénite.
A. L’architecture ganglionnaire est respectée avec une hyperplasie lymphoïde folliculaire. On retrouve la présence de nombreux follicules lymphoïdes secondaires ainsi qu’une expansion des territoires interfolliculaires. B. Au sein des territoires inter-folliculaires, on retrouve une hyperplasie vasculaire et de nombreux polynucléaires éosinophiles. C. On retrouve également la présence de polynucléaires éosinophiles assez nombreux au niveau de la zone du manteau des follicules lymphoïdes secondaires, mais également au sein des centres germinatifs (flèches noires).
La maladie de Kimura est une pathologie bénigne, avec une évolution favorable dans la majorité des cas. Il n’a jamais été décrit de transformation maligne. La rémission post-chirurgicale est fréquente. Des évolutions plus chroniques et indolentes sont néanmoins possibles.
Hyperplasie angio-lymphoïde avec éosinophilie (HALE) ou hémangioendothéliome épithélioïde ?
Il a existé durant de nombreuses décennies une confusion entre la maladie de Kimura et la HALE, alors que les deux pathologies sont bien distinctes (tableau 1). La HALE reste le principal diagnostic différentiel de la maladie de Kimura, les lésions étant de morphologie et de localisation similaires. Les lésions sont vas culaires, angiomateuses, nodulaires ou papuleuses, rosées ou rouge sombre. Les masses sous-cutanées sont recouvertes d’une peau normale et mesurent de 0,2 à 10 cm. La lésion est unique dans 80 % des cas. La localisation préférentielle est ORL, auriculaire ou frontale. Elles ne sont jamais accompagnées d’adénopathies à la différence de la maladie de Kimura, et l’éosinophilie est inconstante.
Sur le plan histologique, il existe à la fois des composantes vasculaire et inflammatoire(13). Les lésions vasculaires consistent en une prolifération de petits vaisseaux sanguins aux cellules endothéliales hypertrophiées, souvent épithélioïdes, faisant hernie dans la lumière et aux noyaux sans atypies ni mitoses anormales. S’y associe un infiltrat dense périvasculaire et interstitiel de lymphocytes, de plasmocytes et d’éosinophiles. Il existe des preuves de l’existence de structures artérielles parmi les veinules, ce qui peut suggérer la présence de shunts artério-veineux.
Les principaux autres diagnostics différentiels regroupent des lymphomes et des maladies systémiques (tableau 2).
LA PHYSIOPATHOLOGIE DE LA MALADIE DE KIMURA RESTE INCONNUE
La principale hypothèse est celle d’une infection, en particulier à Candida albicans, déclenchant un phénomène auto-immun ou conduisant à une réaction d’hypersensibilité de type I (médiée par l’immunoglobuline E). Certaines données suggèrent l’implication des lymphocytes Th2 produisant des cytokines éosinophilopoïétiques, notamment l’interleukine (IL)4, l’IL5 et l’IL13(14,15). Des études ont également montré une élévation du facteur de stimulation des macrophages granulocytaires (GM-CSF), du TNF-alpha et du récepteur soluble de l’IL2(14,15).
La démarche diagnostique
La démarche diagnostique dé pend de la présentation clinique et requiert l’analyse histologique d’un nodule sous-cutané, d’une adénopathie, voire d’une lésion cutanée superficielle.
Malgré l’absence de recommandations sur le bilan étiologique minimal, il doit comprendre les éléments permettant le diagnostic positif et l’élimination des diagnostics différentiels, à savoir une numération formule sanguine, un ionogramme sanguin, un dosage de la créatininémie, un bilan hépatique, une protéine C-réactive, une électrophorèse des protéines sériques, les sérologies virales et une bandelette urinaire à la recherche d’une protéinurie. En fonction de la clinique, pourront être discutés des examens supplémentaires.
Une exploration par un scanner thoraco-abdomino-pelvien voire celle d’un TEP-scanner pour le bilan d’extension initial peut être nécessaire selon la clinique.
Si l’hyperéosinophilie est ancienne et supérieure à 1 000/mm3 , l’exploration de complications de l’hyperéosinophilie doit être réalisée, en particulier la recherche d’une atteinte cardiaque avec électrocardiogramme, échographie cardiaque transthoracique, troponine et NT-pro-BNP, voire l’IRM cardiaque.
Enfin, un déparasitage systématique peut être proposé.
UN TRAITEMENT VARIABLE SELON LA FORME CLINIQUE
Tout comme le bilan étiologique, le traitement de la maladie de Kimura n’est pas codifié. Une surveillance simple est néanmoins souvent admise.Devant une lésion unique volumineuse, une prise en charge chirurgicale avec exérèse complète de la lésion est proposée, éventuellement associée à une radiothérapie localisée. Cette stratégie permet une rémission complète sans rechute dans 30 à 50 % des cas(2,4,16) .
Une corticothérapie par voie orale est recommandée dans les formes plus diffuses ou récidivantes(17,18).
En cas d’atteinte rénale spécifique, l’indication à une corticothérapie à forte dose, de l’ordre de 1 mg/kg/jour, est fréquente, permettant une rémission dans la quasi-totalité des cas, mais des rechutes sont décrites à l’arrêt du traitement dans 40 % des cas(8).
Dans de rares cas, lorsque l’atteinte cutanée est diffuse ou mutilante, ou lorsque l’atteinte rénale est cortico-dépendante ou cortico-résistante, il est possible de proposer le thalidomide ou des immunosuppresseurs comme le mycophénolate mofétil ou la ciclosporine(4). Enfin, des immunothérapies ciblant les éosinophiles et l’inflammation de type Th2 ont été décrites, comme le mépolizumab ciblant l’IL5(19) ou le dupilumab ciblant la sous-unité alpha du récepteur à l’IL4 et l’IL13(20).
* a. Département de médecine interne, Centre de référence maladies rares d’Île-de-France, AP-HP, APHP-CUP, hôpital Cochin, Paris
b. Département de dermatologie, hôpital Saint-Louis, AP-HP, Paris
c. Département de pathologie, Université Paris Cité, Hôpitaux universitaires Necker-Enfants malades et Robert-Debré, APHP, Paris
d. Université de Paris Cité, INSERM, U970, PARCC, Paris
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :